Step #1 prerequists
Step #2 download the latest copy
The latest version can be downloaded from the download page.
Or clone repository
[user@hostname:~] git clone git@forgemia.inra.fr:genotoul-bioinfo/jflow.gitStep #3 extract the archive
[user@hostname:~] tar xzvf jflow_vx.x.tar.gz
[user@hostname:~] cd jflow_vx.xStep #4 configure the application
You must edit the application.properties file to configure the application.
Here under the required parameters, for advanced configuration please see
this page...
[global]
# uncomment and set if not in the PATH, should be version >= 4.4.3
#makeflow = /usr/bin/makeflow
# batch system type: local, condor, sge, moab, cluster, wq, hadoop, mpi-queue
batch_system_type = local
# add these options to all batch submit files
batch_options =
# add these options to limit the number of jobs sumitted in parallel
limit_submission = 100
# on which socket host should run the web server
server_socket_host = 127.0.0.1
# on which socket port should run the web server
server_socket_port = 8080
[storage]
# where should be written the log file
log_file = /path/to/jflow.log
# Where should the pipelines write results, should be accessible
# by all cluster nodes
work_directory = /path/to/work
# Where should the pipelines write temporary files, should be
# accessible by all cluster nodes
tmp_directory = /path/to/tmp
[softwares]
# uncomment and set if not in the PATH
#blastall = /usr/bin/blastall
#formatdb = /usr/bin/formatdb
#sfffile = /usr/bin/sfffile
#fastqc = /usr/bin/fastqc
#runAssembly = /usr/bin/runAssembly
#bwa = /usr/bin/bwa
#samtools = /usr/bin/samtoolsStep #5 test your installation
The application embed one website example, enter the following command lines to run the web server:
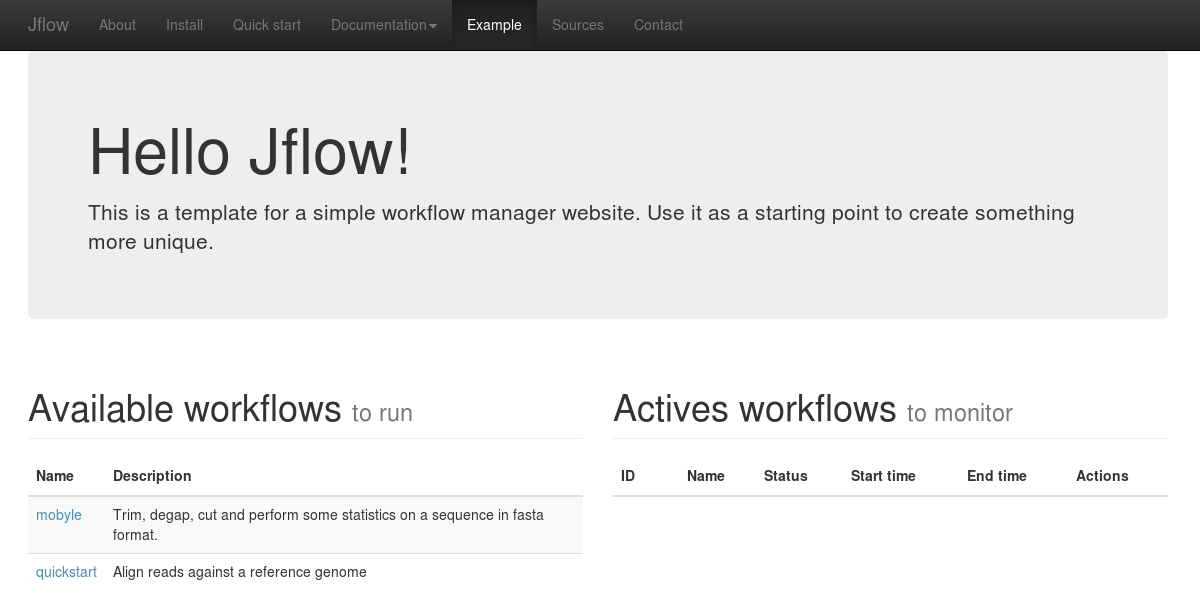
[user@hostname:~] python bin/jflow_server.py --daemonAnd go on the example web page. You should have the following display with the alignment workflow available and no actives workflows.